- +1
識病尋源|從基因編輯技術CRISPR治療失敗看杜氏肌營養(yǎng)不良癥
【編者按】2022年10月,全球唯一一名參與CRISPR基因編輯療法的杜氏肌營養(yǎng)不良癥(DMD)志愿者,在治療過程中死亡。復旦大學基礎醫(yī)學院細胞與遺傳醫(yī)學系研究員楊云龍表示:CRISPR相關技術作為一種高效且廣泛應用的基因編輯方法,是當前研究的熱點。將其應用于臨床,由于涉及到倫理和監(jiān)管問題,也常常成為爭議的焦點。他希望更關注DMD這一遺傳性罕見病本身,DMD目前還沒有治愈方法。“識病尋源”是楊云龍在澎湃科技開設的獨家專欄。
2022年11月10日,《科學》雜志(Science)的新聞欄刊登了一條信息[1]:一名27歲杜氏肌營養(yǎng)不良癥患者在一項新型基因編輯試驗中死亡。雖然信息披露尚不充分,但許多科學家認為,這名患者確實通過病毒輸注,接受了基于CRISPR的治療。
根據(jù)ClinicalTrials.gov上的注冊信息(NCT05514249),該臨床試驗僅有一名參與者。受試者2022年8月底在美國馬薩諸塞大學醫(yī)學院接受了靜脈輸注,約6周后去世。CRISPR相關技術作為一種高效且廣泛應用的基因編輯方法,是當前研究的熱點。將其應用于臨床,由于涉及到倫理和監(jiān)管問題,也常常成為爭議的焦點。由于基因編輯的話題性,大量的新聞網(wǎng)站報道了該事件。但新聞報道中并未對受試者患有的杜氏肌營養(yǎng)不良癥作詳盡解釋。例如,在美國廣播公司(American Broadcasting Company, ABC)的報道中,僅有一句話描述該病:“這種罕見的遺傳性肌肉萎縮疾病是由產生一種叫做肌營養(yǎng)不良蛋白的基因突變引起”。
杜氏肌營養(yǎng)不良癥(Duchenne Muscular Dystrophy, DMD)得名于紀堯姆·本杰明·阿曼德·杜興(Guillaume Benjamin Amand Duchenne, 1806-1875)。他是200年前的法國神經生物學家,進行了肌肉電刺激的開創(chuàng)性研究。杜興不僅嘗試用電治療肌肉問題,還將電作為研究解剖結構和生理學的研究手段。他發(fā)明了“杜興機”—一種用電刺激肌肉的便攜裝置,極大地豐富了電生理學研究。使用該裝置,他發(fā)現(xiàn)假笑不涉及眼睛周圍的肌肉,而發(fā)自內心的微笑涉及眼周和嘴部肌肉。如今,這種需要調動眼輪匝肌的真誠笑容被稱為“杜興笑”。實驗過程中拍攝的電刺激面部肌肉的照片令人印象深刻,廣為傳播。部分原始照片現(xiàn)已被收入美國紐約的現(xiàn)代藝術博物館(Museum of Modern Art, MoMA)
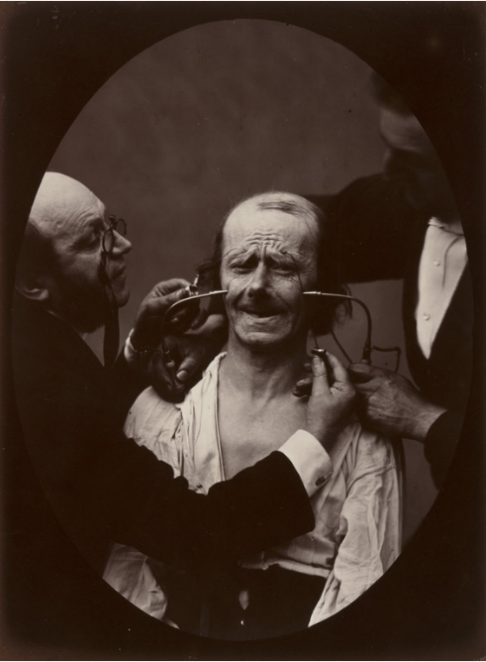
杜興用電刺激受試者面部肌肉的照片,1856。來源:MoMA館藏。
1842年起,杜興在巴黎開展肌營養(yǎng)不良和其他神經肌肉疾病的研究,嘗試用電刺激觸發(fā)特定肌肉運動,并詳細描述了幾類神經肌肉疾病。在杜興之前,DMD的病例已經被零星地描述過,如1830年,英國著名醫(yī)生查爾斯·貝爾(Charles Bell, 1774-1842)描述了一名肌肉進行性麻痹導致下肢癱瘓的18歲男子,從10歲起大腿無力,不能站立[2]。1847年理查德·帕特里奇(Richard Partridge,1805–1873)第一次對DMD患者進行病理尸檢,發(fā)現(xiàn)“肌肉出現(xiàn)脂肪變性,小腿變性程度高于上肢,神經和肌腱都沒有發(fā)生變化”[3]。1851年,愛德華·梅里恩(Edward Meryon,1807–1880)首次對DMD進行了明確的臨床和病理描述,他向倫敦皇家醫(yī)學和外科學會提交了論文,描述患者尸檢結果:“在顯微鏡下檢查肌肉組織時,發(fā)現(xiàn)條紋狀纖維被完全破壞,肌節(jié)成分彌散,在許多地方轉化為脂滴和顆粒物,肌纖維膜被破壞”[4]。梅里恩最初認為DMD患者是由于脊髓問題導致,但經過仔細檢查,他發(fā)現(xiàn)患者神經組織是完整的,觀察到的唯一結構變化是肌纖維改變,脂肪填充了肌肉纖維鞘。從1851年到1870年,梅里恩一直對這一疾病開展研究,可能是全面總結DMD的第一人。杜興則首次拍攝了DMD患者的照片,1862年發(fā)表在照片集《病理攝影集》(Album de photographes pathologiques)中[5],這也是首次出版的臨床影像資料的書籍。更進一步,杜興發(fā)明了一種肌肉活檢針,于1864年應用,對一名DMD患者肌肉進行了活檢觀察。杜興高質量地確定了DMD的關鍵病理結果,并在1868年總結了該病的特征。隨后,神經科學家威廉·理查德·高爾斯爵士(Sir William Richard Gowers,1845–1915)在觀察了多個家系后得出結論,DMD是一種具有明顯男性偏好的早發(fā)性遺傳疾病,其病因來自卵子。大多數(shù)病例6歲前發(fā)病,由于腿部肌肉乏力,具有特征性的站起姿勢。高爾斯嘗試過多種療法,包括杜興的電刺激療法,未觀察到療效[6]。在確定DMD的疾病特征、病理改變的過程中,杜興、高爾斯等人做出了大量的貢獻。2003年,《神經病學年鑒》(Annals of Neurology)期刊主編Kenneth Tyler教授對DMD的早期探索歷史進行了總結,并在美國神經病學年會上報告[7]。

杜興為他第一位DMD患者拍攝的影像資料。來源:[4]
進入20世紀,DMD研究一度陷入沉寂。直到進入分子時代,大量的遺傳學研究和分子生物學研究聚焦于該疾病,極大地豐富了我們對DMD的認知。通過流行病學調查,我們知道杜氏肌營養(yǎng)不良(DMD)是一種X連鎖隱性遺傳病,發(fā)病率在男性嬰兒中約十萬分之10,屬于發(fā)病率較高的遺傳病。另一類稱為貝氏肌營養(yǎng)不良(Becker muscular dystrophy, BMD)與其發(fā)病機理類似,發(fā)病率為約十萬分之8。它們在女性中極為罕見。
究其病因,DMD和BMD是由肌細胞中一個重要蛋白Dystrophin遺傳改變,導致結構及功能缺失所致。編碼蛋白的基因體量巨大,是人類基因組中最大的基因之一。具有79個外顯子,翻譯出的蛋白質量有427kDa。Dystrophin通過兩端的結構域將細胞骨架蛋白F-actin與細胞外基質連接起來。此外,Dystrophin還結合肌膜、細胞骨架、通道蛋白以及信號蛋白,形成復雜的復合物,并介導一系列功能。目前對這些功能的理解還不充分。在DMD患者中,Dystrophin被截斷,導致細胞骨架無法連接到細胞外基質。在BMD中,Dystrophin的關鍵結構域尚存,但結構不完整,所以病情較輕。遺傳突變是導致Dystrophin丟失或損傷的直接原因。迄今,已在DMD和BMD患者中發(fā)現(xiàn)了數(shù)千種不同類型的突變。大多數(shù)的突變發(fā)生在熱點區(qū)域,即3號-9號外顯子以及45號-55號外顯子區(qū)域。
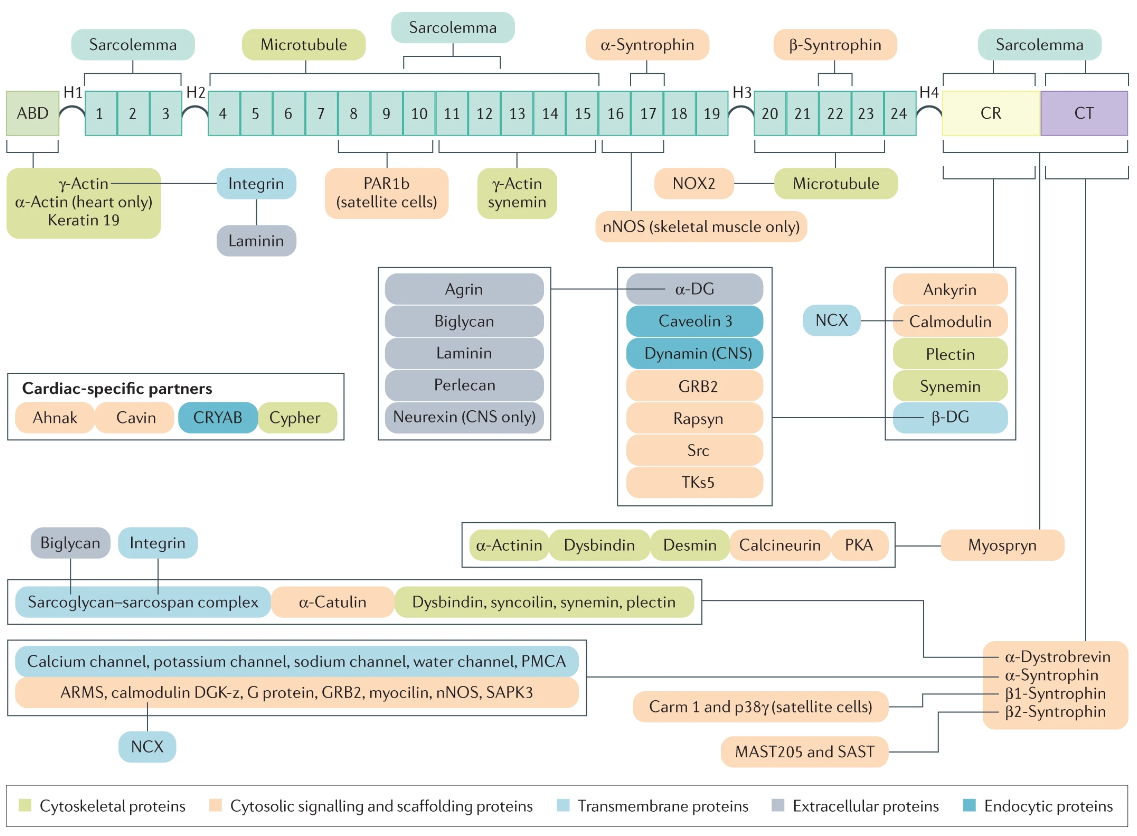
Dystrophin可以和多種蛋白結合,其缺失廣泛影響肌細胞功能。來源:[8]
Dystrophin發(fā)生問題后,以其為中心的復合物會解體,肌細胞結構完整性、肌肉收縮能力、肌細胞的信號傳導都會受到影響。體現(xiàn)在肌細胞上,就是肌膜易受收縮損傷、細胞被自由基損傷、鈣離子超載、再生受阻等。這些受損的肌細胞會被炎癥細胞識別并清除。在疾病早期,受損肌肉尚可通過再生修復。但在晚期,由于再生能力降低和纖維化信號上調,肌細胞被脂肪組織和纖維化組織取代,進而影響患者運動能力。
DMD目前沒有治愈方法,但合理治療可緩解疾病進展,減輕患者負擔。在疾病診斷方面,若發(fā)現(xiàn)男孩有癥狀,要對患者家庭開展基因咨詢,協(xié)助診斷。這有助于分型并指導未來治療。在治療方面,目前的標準方案是:DMD患兒在其運動發(fā)育停止時(一般在4-5歲)使用糖皮質激素,并在整個生命周期內持續(xù)治療,這主要是由于糖皮質激素可促進肌細胞增殖同時抑制炎癥。該療法可延緩病情,增進患者生存。同時,對癥的多學科治療和高質量的護理也可改善生活質量和壽命。這包括醫(yī)師、護士、康復師、營養(yǎng)師、心理醫(yī)生等的共同合作。對于DMD的診療方案,2018年發(fā)表于Lancet Neurology的指南做了最為完善的總結 [9-11]。
由于DMD的不可治愈,在科研領域,科學家仍在不斷尋找肌營養(yǎng)不良的療法,確認Dystrophin缺失是病因后,一個樸素的“治本”的想法就是能否令肌細胞重新表達這一蛋白。由于編碼蛋白的基因體量過大,直接導入全長基因并不現(xiàn)實。于是科學家們開發(fā)了一系列新療法,主要包括基因療法和細胞療法。具體可分為以下幾類:
終止密碼子通讀:有5%-7%的DMD病例,是由于基因突變形成了終止密碼子,也就是無義突變。該突變導致Dystrophin合成提前終止。若能強制要求蛋白繼續(xù)合成,就可能得到一個全長的蛋白,從而治療疾病。一種小分子藥物Ataluren可以誘導核糖體在遇到無義突變時繼續(xù)向下翻譯。在臨床試驗中,該藥物未能達到主要終點,但顯示了一定的治療趨勢,且安全性良好。因此得到了歐洲藥監(jiān)局的條件性使用批準。
外顯子跳躍:蛋白表達需要先轉錄為mRNA,通過剪接將多個外顯子拼在一起。某些特定的突變可導致蛋白合成提前終止。若能跳過該突變所在的外顯子區(qū)域,產生一個不是全長的蛋白。雖然仍不及健康蛋白,但可顯著減輕病情。這一方法利用寡核苷酸片段改變剪接過程,體積小,易于遞送,安全性好。該方法針對不同突變要設計不同的寡核苷酸藥物,目前的藥物針對突變最多的群體,如外顯子44、45、51、53等,需要重復治療。其中部分藥物已獲美國藥監(jiān)局的條件性使用批準。
表達微型蛋白:使用腺相關病毒(Adeno-associated virus, AAV)可遞送cDNA,令感染的組織表達特定蛋白。該方法也是其他基因療法中常見方案。但在DMD中,由于AAV容量有限,無法運載編碼Dystrophin的載體。于是科學家提出了僅將最關鍵結構域重組為微型蛋白,再用AAV導入的方法。臨床試驗結果證實,微型蛋白在受試者肌細胞中成功表達,但目前還不知道對疾病的改善情況。需要注意的是,AAV可能導致患者出現(xiàn)嚴重的免疫反應。即使對患者進行了預篩選和免疫抑制,仍有患者出現(xiàn)了嚴重不良事件。 2021年12月,輝瑞公司在一例受試者報告死亡后,暫停臨床試驗,目前正在恢復中。
基因組編輯技術:如果能夠對基因組直接進行靶向修飾,糾正突變,或者將導致蛋白丟失的突變刪除或替換,是非常有吸引力的方法。通過設計引導RNA,引入CRISPR系統(tǒng),可以做到在基因組特定位點刪除外顯子、取消剪接位點等。該方法在動物實驗中已獲成功。但由于基因編輯的脫靶風險和對其中的蛋白或病毒載體產生免疫反應的風險,基因組編輯尚未進入臨床。開篇提及的新聞報道中,死亡的患者可能參加了這一療法的臨床研究。
干細胞移植:將帶有正常基因的干細胞移植,修復肌肉。雖然該方法有吸引力,但肌肉組織的體量對該方法提出了挑戰(zhàn)。臨床試驗也發(fā)現(xiàn),僅在局部注射的位置旁有一些恢復。該方法目前未獲批準。
類似蛋白替代:Dystrophin有一種類似蛋白稱為Utrophin,若上調其表達可產生一定的代償效應。有臨床試驗研究了能夠上調Utrophin的藥物,但未觀察到治療效果。
除了這些“治本”的療法,還有許多藥理學藥物在臨床試驗中,或可通過減少纖維化、減輕炎癥,擴張血管、保護線粒體功能、靶向肌肉調節(jié)蛋白等不同機制,可能緩解病情[12]。
從患者的角度看來,多種療法帶來了治愈疾病的希望,但治療方案與患者的適配性仍有待考量,且方法之間可能互斥。綜合比較,表達微型蛋白是一種普適的方法,但其仍存在安全隱患。其不良反應可能主要由AAV介導,涉及制造過程中的諸多因素,如劑量、AAV類型、轉入的載體、患者是否已有抗體等。開篇提到的新聞報道中,患者應該是選擇了基于CRISPR的基因組編輯技術。報道體現(xiàn)了公眾對以CRISPR為代表的基因療法的擔憂。在該試驗更多信息披露之前,更重要的可能是提高公眾對疾病和各類療法的深入理解。更進一步,患者和患者家屬通過理解相關研究,決定治療方案,決定是否參與前沿療法的臨床研究,需要勇氣、知識、智慧和積極的人生觀。而對于患有DMD這樣的罕見病患者,在某種程度上,積極的人生觀要比新穎的療法更為重要。
(作者楊云龍,系復旦大學基礎醫(yī)學院細胞與遺傳醫(yī)學系研究員、副主任。疾病不斷地改變著每個人的人生軌跡。但除了醫(yī)生與醫(yī)學研究者,人們很少有機會了解各式各樣的疾病。“識病尋源”專欄將以一文一病的形式,介紹對疾病的認識進程,疾病的病因及其治療。跟隨醫(yī)學科學的進步,理解現(xiàn)代醫(yī)學。)
參考文獻
1. https://www.science.org/content/article/news-glance-new-antibiotic-covid-19-antarctica-and-venus-mission-deferred. doi: 10.1126/science.adf7363
2. Bell, C., The nervous system of the human body : embracing the papers delivered to the Royal Society on the subject of the nerves. 1830, London: Longman, Rees, Orme, Brown, and Green. xxiii, 238, clxxvi p., 9 leaves of plates.
3. Patridge R. Fatty degeneration of muscle. Med Times Gaz 1847; 5: 944.)
4. Meryon, E., On Granular and Fatty Degeneration of the Voluntary Muscles. Med Chir Trans, 1852. 35: p. 73-84 1.
5. Duchenne GBA. Album de photographes pathologiques. Paris: Bailliere; 1862.
6. Gowers WR. Clinical lecture on pseudo-hypertrophic muscular paralysis. Lancet 1879; 2: 1–2, 37–39, 73–75, 113–116.
7. Tyler, K.L., Origins and early descriptions of "Duchenne muscular dystrophy". Muscle Nerve, 2003. 28(4): p. 402-22.
8. Duan, D., et al., Duchenne muscular dystrophy. Nat Rev Dis Primers, 2021. 7(1): p. 13.
9. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol, 2018. 17(3): p. 251-267.
10. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol, 2018. 17(4): p. 347-361.
11. Birnkrant, D.J., et al., Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol, 2018. 17(5): p. 445-455.
12. Markati, T., et al., Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol, 2022. 21(9): p. 814-829.



- 報料熱線: 021-962866
- 報料郵箱: news@thepaper.cn
互聯(lián)網(wǎng)新聞信息服務許可證:31120170006
增值電信業(yè)務經營許可證:滬B2-2017116
? 2014-2025 上海東方報業(yè)有限公司

