- +1
《自然》:AlphaFold和AI蛋白質(zhì)折疊革命終局如何
在過去的半年里,AlphaFold狂熱席卷了生命科學領域。“我參加的每次會議,人們都在說‘為什么不使用AlphaFold?’。”倫敦大學學院的計算生物學家Christine Orengo說。
當?shù)貢r間4月13日,《Nature》發(fā)文總結了自AlphaFold出現(xiàn)以來,在生命科學領域產(chǎn)生了哪些令人矚目的成果,以及伴隨著AlphaFold的發(fā)展,又給生命科學帶來哪些新的可能性。最后,AlphaFold有何限制,其將去向何方?
以下為記者對《What's next for AlphaFold and the AI protein-folding revolution》原文的編譯:
在某些情況下,人工智能節(jié)省了科學家的時間,甚至在一些情況下,它使以前不可想象或非常不切實際的研究成為可能。同時它也有局限性,一些科學家發(fā)現(xiàn)它的預測對他們的工作來說太不可靠了。但實驗仍然如火如荼。
即使是開發(fā)AlphaFold的人也在努力跟上它在從藥物發(fā)現(xiàn)和蛋白質(zhì)設計到復雜生命起源等領域的使用。“我醒來后在Twitter上輸入AlphaFold,”DeepMind的AlphaFold團隊負責人John Jumper說,“看到一切都有相當神奇之感。”
“AlphaFold改變了游戲規(guī)則。”Baker說。“這就像一場地震。你可以在任何地方看到它,”以色列耶路撒冷希伯來大學的計算結構生物學家Ora Schueler-Furman說,他正在使用AlphaFold來模擬蛋白質(zhì)相互作用。
AlphaFold在2020年12月引起了轟動,當時它主導了一場名為“蛋白質(zhì)結構預測關鍵評估”(Critical Assessment of Protein Structure Prediction,CASP)的比賽。該競賽每兩年舉行一次,旨在衡量生物學最大挑戰(zhàn)之一的進展:僅從蛋白質(zhì)的氨基酸序列中確定蛋白質(zhì)的3D形狀。計算機軟件根據(jù)X射線晶體學或低溫電子顯微鏡(cryo-EM)等實驗方法,通過向蛋白質(zhì)發(fā)射X射線或電子束以建立它們的圖像形狀。
AlphaFold的2020版是該軟件的第二版。Jumper說,它還贏得了2018年的 CASP,但其早期的努力大多不足以代替實驗確定的結構,而AlphaFold2的預測平均而言與經(jīng)驗結構已相當。
此前尚不清楚DeepMind何時會發(fā)布可廣泛使用的軟件,因此研究人員利用Jumper的公開演講中的信息以及他們自己的見解,開發(fā)了自己的AI工具,稱為RoseTTAFold。
此后在2021年7月15日,描述RoseTTAFold和AlphaFold2的論文出現(xiàn)了,還有免費提供的開源代碼和專家運行他們自己工具版本所需的其他信息。一周后,DeepMind宣布它已經(jīng)使用AlphaFold預測了人類制造的幾乎所有蛋白質(zhì)的結構,以及其他20種廣泛研究的生物體的整個“蛋白質(zhì)組”,例如小鼠和大腸桿菌數(shù)量超過36.5萬的總結構。DeepMind還將這些信息公開發(fā)布到由EMBL的歐洲生物信息學研究所維護的數(shù)據(jù)庫中(EMBL-EBI)。此后,該數(shù)據(jù)庫已增長至近一百萬個結構。
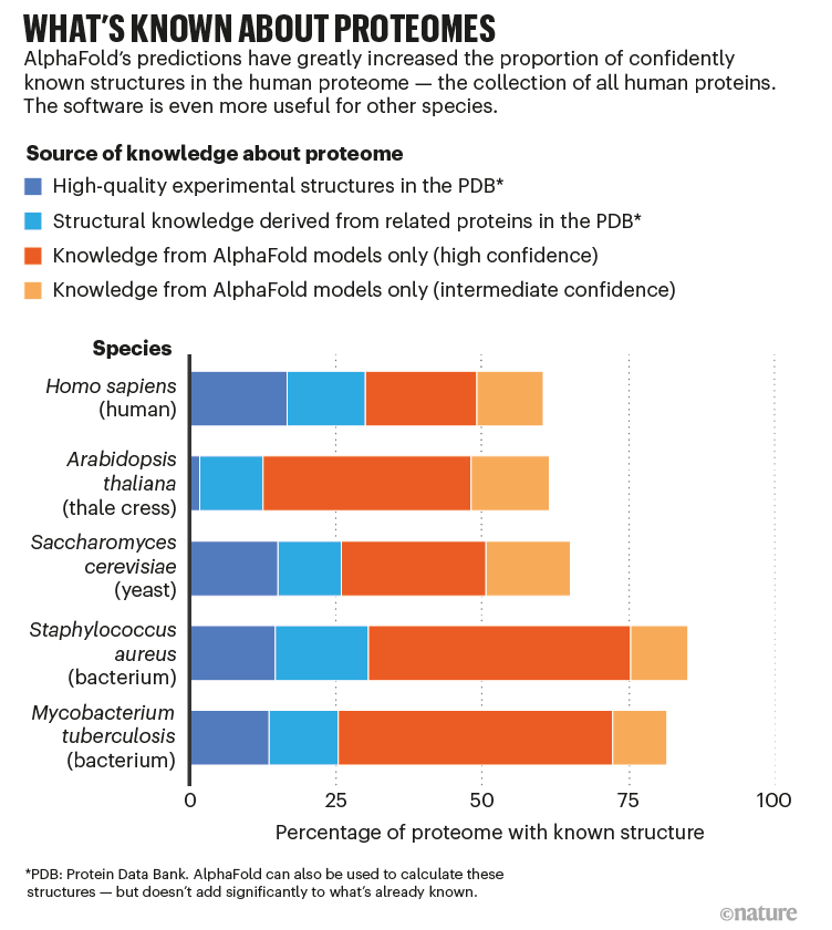
今年,DeepMind計劃發(fā)布總計超過1億個結構預測。這幾乎是所有已知蛋白質(zhì)的一半,是蛋白質(zhì)數(shù)據(jù)庫 (PDB)結構庫中實驗確定的蛋白質(zhì)數(shù)量的數(shù)百倍。
AlphaFold部署了深度學習神經(jīng)網(wǎng)絡(一種受大腦神經(jīng)線路啟發(fā)的計算架構,可識別數(shù)據(jù)中的模式),它目前已經(jīng)接受了蛋白質(zhì)數(shù)據(jù)庫和其他數(shù)據(jù)庫中數(shù)十萬個實驗確定的蛋白質(zhì)結構和序列的訓練。當面對一個新序列時,它將首先在數(shù)據(jù)庫中尋找相關序列,這些序列可以識別出傾向于一起進化的氨基酸,表明它們在3D空間中很接近。而現(xiàn)有相關蛋白質(zhì)的結構可以提供另一種估計新序列中氨基酸對之間距離的方法。
AlphaFold在嘗試對氨基酸的3D位置進行建模時來回迭代來自這些平行軌跡的線索,并不斷更新其估計值。專家表示,該軟件在機器學習研究中的新思想應用似乎是AlphaFold如此出色的原因——特別是,它使用一種稱為“注意力”(attention)的人工智能機制來確定哪些氨基酸連接在任何時候?qū)ζ淙蝿兆钪匾?/p>
該網(wǎng)絡對相關蛋白質(zhì)序列信息的依賴意味著AlphaFold存在一些局限性。即它并非旨在預測突變(例如引起疾病的突變)對蛋白質(zhì)形狀的影響,它也沒有被訓練來確定在其他相互作用的蛋白質(zhì)或藥物等分子存在的情況下蛋白質(zhì)如何改變形狀。但它的模型附帶的分數(shù)可以衡量網(wǎng)絡對其預測蛋白質(zhì)每個氨基酸單元的信心,研究人員正在調(diào)整AlphaFold的代碼以擴展其功能。
據(jù)DeepMind稱,到目前為止,已有超過40萬人使用了EMBL-EBI的AlphaFold數(shù)據(jù)庫。
用AlphaFold求解蛋白質(zhì)結構
AlphaFold解析結構的能力已經(jīng)給生物學家留下了深刻的印象。“根據(jù)我目前所見,我非常信任AlphaFold。”丹麥奧胡斯大學(Aarhus University)的結構生物學家Thomas Boesen說。該軟件已成功預測了Boesen中心已確定但尚未發(fā)表的蛋白質(zhì)形狀,“這對我來說是一個很大的驗證。”
Boesen和奧胡斯微生物生態(tài)學家Tina ?antl-Temkiv正在使用AlphaFold來模擬促進冰形成的細菌蛋白質(zhì)結構——這可能有助于云中冰的冷卻效應。
斯德哥爾摩大學的蛋白質(zhì)生物信息學家Arne Elofsson說,只要一種蛋白質(zhì)卷曲成一個明確定義的3D形狀,而且不需要全部如此,AlphaFold的預測就很難被擊敗。“這是一種一鍵式解決方案,可能是你將獲得的最佳模型。”
Elofsson說,在AlphaFold不太自信的地方,“它非常擅長告訴你什么時候它不起作用”。在這種情況下,預測的結構可能類似于浮動的意大利面條線(如下圖)。這通常對應于缺乏確定形狀的蛋白質(zhì)區(qū)域,這種本質(zhì)上無序的區(qū)域——約占人類蛋白質(zhì)組的三分之一,可能只有在存在另一種分子(如信號伙伴)時才能得到很好的定義。
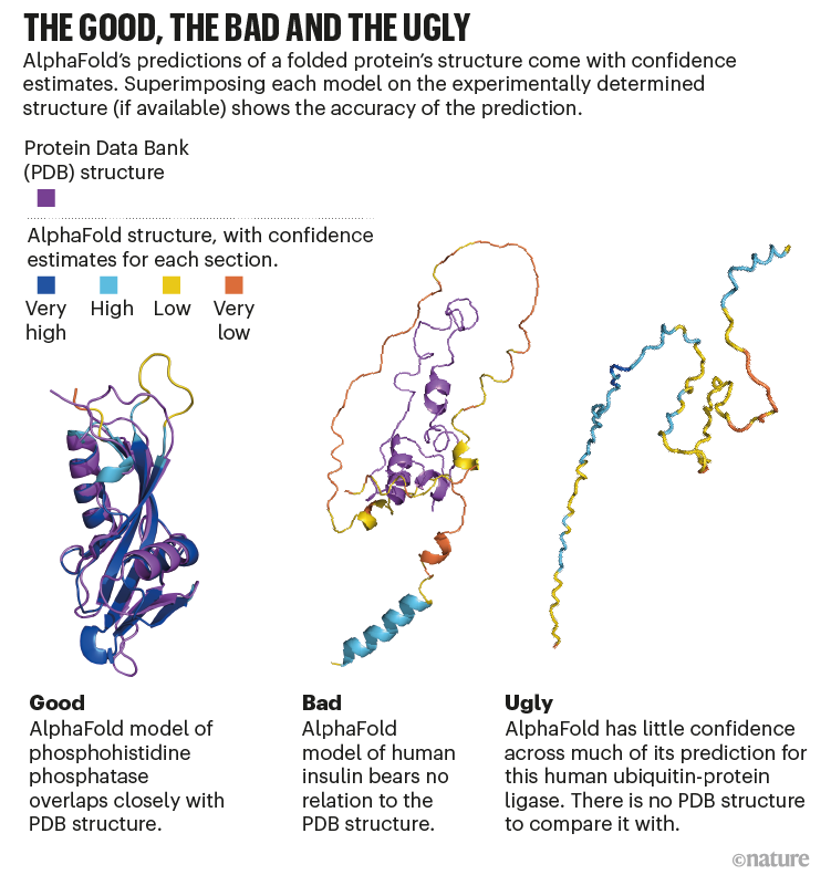
倫敦癌癥研究所的計算生物學家Norman Davey表示,AlphaFold識別疾病的能力已經(jīng)改變了他研究這些區(qū)域特性的工作,“我們的預測質(zhì)量立即有了巨大的提高,而我們沒有付出任何努力。”
AlphaFold轉儲到EMBL-EBI數(shù)據(jù)庫中的蛋白質(zhì)結構數(shù)據(jù)也立即投入使用。Orengo的團隊正在搜索它以識別新的蛋白質(zhì)種類(沒有通過實驗驗證它們),并且已經(jīng)發(fā)現(xiàn)了數(shù)百甚至數(shù)千個潛在的新蛋白質(zhì)家族,擴大了科學家對蛋白質(zhì)外觀和功能的了解。
在另一項努力中,該團隊正在搜索從海洋和廢水中采集的DNA序列數(shù)據(jù)庫,以嘗試識別新的食用塑料酶。使用AlphaFold快速模擬近似的數(shù)千種蛋白質(zhì)的結構,研究人員希望更好地了解酶如何進化以分解塑料,并有何潛在改進可能。
“對于進化研究來說,將任何蛋白質(zhì)編碼基因序列轉化為可靠結構的能力是特別重要的。”哈佛大學的進化生物學家Sergey Ovchinnikov認為,研究人員通過比較基因序列以確定生物及其基因在物種間的相關性。但對于遠緣相關的基因,比較可能無法找到進化親屬,因為序列發(fā)生了很大變化。那么通過比較蛋白質(zhì)結構——其變化往往不如基因序列快——研究人員或許能夠發(fā)現(xiàn)被忽視的古老關系。“這為研究蛋白質(zhì)進化和生命起源提供了一個絕佳的機會。”蘇黎世瑞士聯(lián)邦理工學院的計算生物學家Pedro Beltrao表示。
為了驗證這個想法,首爾國立大學的計算生物學家Martin Steinegger和他的同事領導的一個團隊使用他們開發(fā)的“Foldseek”工具來尋找SARS-CoV-2的RNA復制酶的親屬(一種導致COVID-19的病毒)。在EMBL-EBI的AlphaFold數(shù)據(jù)庫中,這項搜索發(fā)現(xiàn)了以前未知的可能遠古親屬:真核生物中的蛋白質(zhì),包括粘菌,它們的3D結構類似于稱為逆轉錄酶的酶。病毒如HIV使用逆轉錄酶將RNA復制到DNA中,盡管在遺傳基因序列水平幾乎沒有相似性。
作為實驗助理的AlphaFold
對于想要確定特定蛋白質(zhì)的詳細結構的科學家來說,用AlphaFold預測不一定是直接解決方案。比起這個,它能提供的是一個可以通過實驗驗證或改進的初始近似值,這本身有助于理解實驗數(shù)據(jù)。例如,來自X射線晶體學的原始數(shù)據(jù)顯示為衍射X射線的圖案,科學家通常需要對蛋白質(zhì)結構進行初步猜測才能解釋這些模式。
“以前,他們經(jīng)常將來自蛋白質(zhì)數(shù)據(jù)庫中相關蛋白質(zhì)的信息拼湊起來或使用實驗方法。”英國劍橋大學的結構生物學家Randy Read說,他的實驗室專門研究其中一些方法。現(xiàn)在,AlphaFold的預測使得大多數(shù)X射線模式不需要這種方法。Read說,他的實驗室正在努力在實驗模型中更好地利用AlphaFold,“我們完全重新調(diào)整了研究重點。”
Read和其他研究人員已經(jīng)使用AlphaFold從X射線數(shù)據(jù)中確定晶體結構,這些數(shù)據(jù)在沒有足夠的起始模型的情況下是無法解釋的。“人們正在解決多年來一直沒有解決的結構。”Read實驗室的前博士后Claudia Millán Nebot說。她預計會看到大量新的蛋白質(zhì)結構提交給蛋白質(zhì)數(shù)據(jù)庫,“這在很大程度上是因為AlphaFold”。
專門從事冷凍電鏡研究(捕捉快速冷凍蛋白質(zhì)的照片)的實驗室也是如此。在某些情況下,AlphaFold的模型準確地預測了稱為G蛋白偶聯(lián)受體(GPCR)的蛋白質(zhì)獨特特征,這些蛋白質(zhì)是重要的藥物靶標。“它似乎非常適合生成第一個模型,然后我們用一些實驗數(shù)據(jù)對其進行改進,”北卡羅來納大學教堂山分校的結構生物學家和藥理學家Bryan Roth說,“這為我們節(jié)省了一些時間。”
但AlphaFold并不總是那么準確,Roth補充說,在他的實驗室已經(jīng)解決但尚未發(fā)表的幾十個GPCR結構中,“大約有一半的時間,AlphaFold結構相當好,而有一半的時間它們或多或少對我們的目的毫無用處。在某些情況下,AlphaFold以高置信度標記預測,但實驗結構表明它是錯誤的。即使軟件做對了,它也無法模擬蛋白質(zhì)與藥物或其他小分子(配體)結合時的外觀,這會大大改變結構。”這樣的警告讓Roth想知道AlphaFold對藥物發(fā)現(xiàn)有多大用處。
在藥物發(fā)現(xiàn)工作中越來越普遍使用計算對接軟件(computational-docking software)來篩選數(shù)十億個小分子以找到一些可能與蛋白質(zhì)結合的小分子(這表明它們可以制造有用的藥物)。Roth現(xiàn)在正與加州大學舊金山分校的藥物化學家Brian Shoichet合作,以了解AlphaFold的預測與通過實驗確定的結構相比如何。
Shoichet說,他們正將工作限制在AlphaFold的預測與實驗結構相吻合的蛋白質(zhì)上。但即使在這些情況下,對接軟件也會為實驗結構和AlphaFold提供不同的藥物命中率,這表明微小的差異可能很重要。“這并不意味著我們不會找到新的配體,我們只會找到不同的配體。”Shoichet說。他的團隊現(xiàn)在正在合成使用AlphaFold結構鑒定的潛在藥物,并在實驗室中測試它們的活性。
AlphaFold可以幫助藥物發(fā)現(xiàn)?學者:帶著批判性的樂觀
Shoichet說,制藥公司和生物技術公司的研究人員對AlphaFold幫助藥物發(fā)現(xiàn)的潛力感到興奮。“批判性的樂觀就是我的描述。”2021年11月,DeepMind推出了自己的衍生產(chǎn)品IsoMorphic Labs,旨在將AlphaFold和其他AI工具應用于藥物發(fā)現(xiàn),但該公司對其具體計劃只字未提。
Karen Akinsanya領導著Schr?dinger的治療開發(fā),Schr?dinger是一家總部位于紐約市的藥物發(fā)現(xiàn)公司,該公司也發(fā)布化學模擬軟件。Akinsanya說,她和同事已經(jīng)在虛擬屏幕和候選藥物的化合物設計中成功地使用AlphaFold結構,包括G蛋白偶聯(lián)受體(GPCR)。她發(fā)現(xiàn),就像實驗結構一樣,需要額外的軟件來獲取氨基酸側鏈或單個氫原子可能所在位置的詳細信息。一旦完成,AlphaFold結構已被證明在某些情況下足以指導藥物發(fā)現(xiàn)。
“很難說‘這是靈丹妙藥’;因為它可以令人驚訝的為一個結構做得很好,但它顯然不是非常適用于所有結構。”Akinsanya說。她和同事發(fā)現(xiàn),AlphaFold的準確度預測并不能顯示一個結構是否對以后的藥物篩選有用。Akinsanya認為,AlphaFold結構永遠不會完全取代藥物發(fā)現(xiàn)中的實驗性結構,但他們可能會通過補充實驗方法來加速這一過程。
對AlphaFold感到好奇的藥物開發(fā)商在1月份收到了好消息,當時DeepMind取消了對其用于商業(yè)應用的關鍵限制。當該公司在2021年7月發(fā)布AlphaFold的代碼時,它規(guī)定運行AlphaFold神經(jīng)網(wǎng)絡所需的參數(shù)或權重。這是在數(shù)十萬個蛋白質(zhì)結構和序列上訓練的最終結果,僅用于非商業(yè)用途。Akinsanya表示,這對行業(yè)中的一些人來說是一個瓶頸,當DeepMind改變策略時出現(xiàn)了一股“興奮的浪潮”。(RoseTTAFold也有類似的限制,其開發(fā)人員之一Ovchinnikov說,但下一個版本將完全開源。)
人工智能工具不僅改變了科學家確定蛋白質(zhì)外觀的方式,一些研究人員正在使用它們來制造全新的蛋白質(zhì)。“深度學習正在徹底改變我們小組中蛋白質(zhì)設計的方式。”華盛頓大學的生物化學家、蛋白質(zhì)設計和預測其結構領域的領導者David Baker說。Baker的團隊與計算化學家Minkyung Baek一起領導了開發(fā)RoseTTAFold的工作。
Baker的團隊讓AlphaFold和RoseTTAFold能夠產(chǎn)生新蛋白質(zhì)“幻覺”。研究人員已經(jīng)改變了人工智能代碼,因此,給定氨基酸的隨機序列,軟件將對其進行優(yōu)化,直到它們類似于神經(jīng)網(wǎng)絡識別為蛋白質(zhì)的東西。
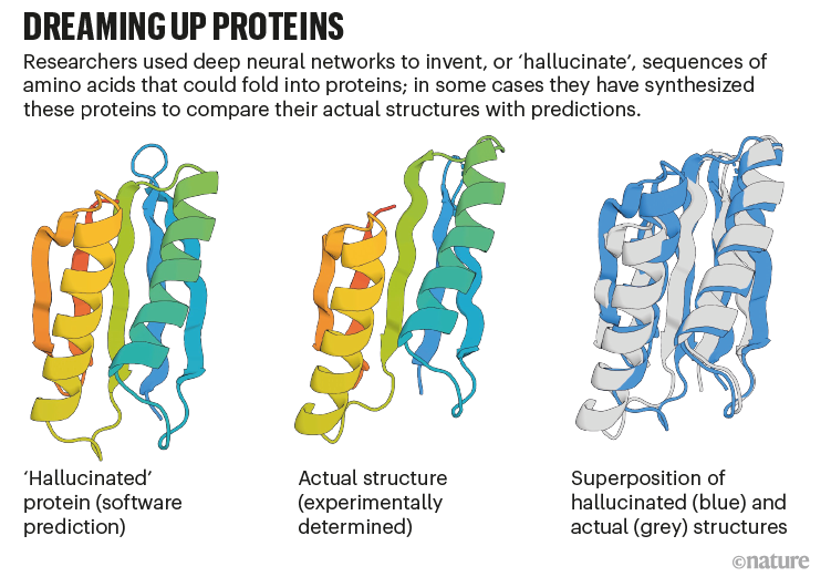
2021年12月,Baker和他的同事報告說,在細菌中表達了129種這些幻覺蛋白,并發(fā)現(xiàn)其中約五分之一折疊成類似于其預測形狀的東西。“這確實是第一次證明你可以使用這些網(wǎng)絡設計蛋白質(zhì)。”Baker說。他的團隊現(xiàn)在正在使用這種方法來設計做有用事情的蛋白質(zhì),例如催化特定的化學反應,方法是指定負責所需功能的氨基酸,并讓AI“夢想”其余部分。
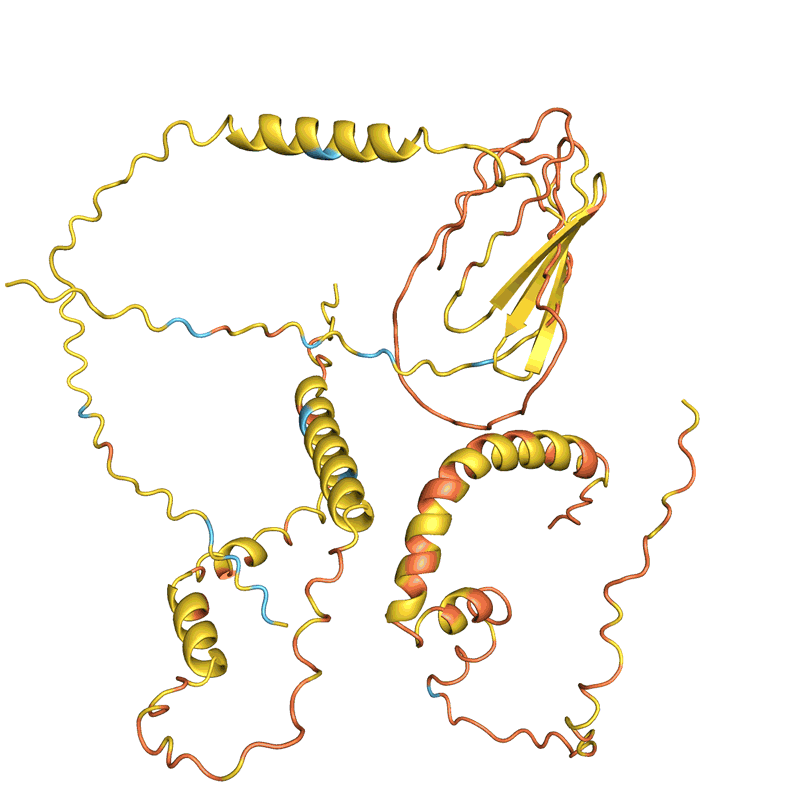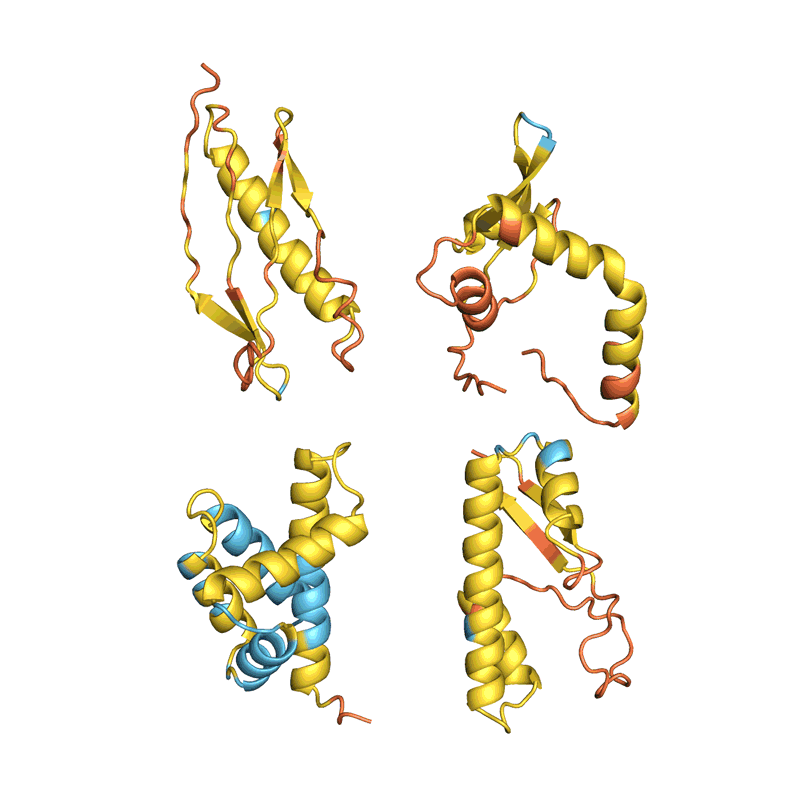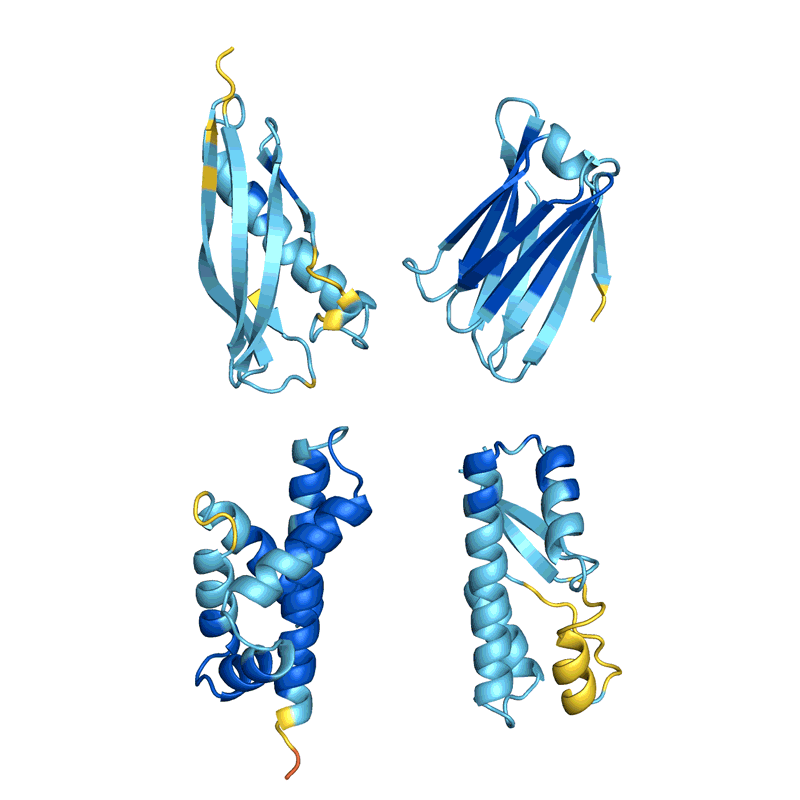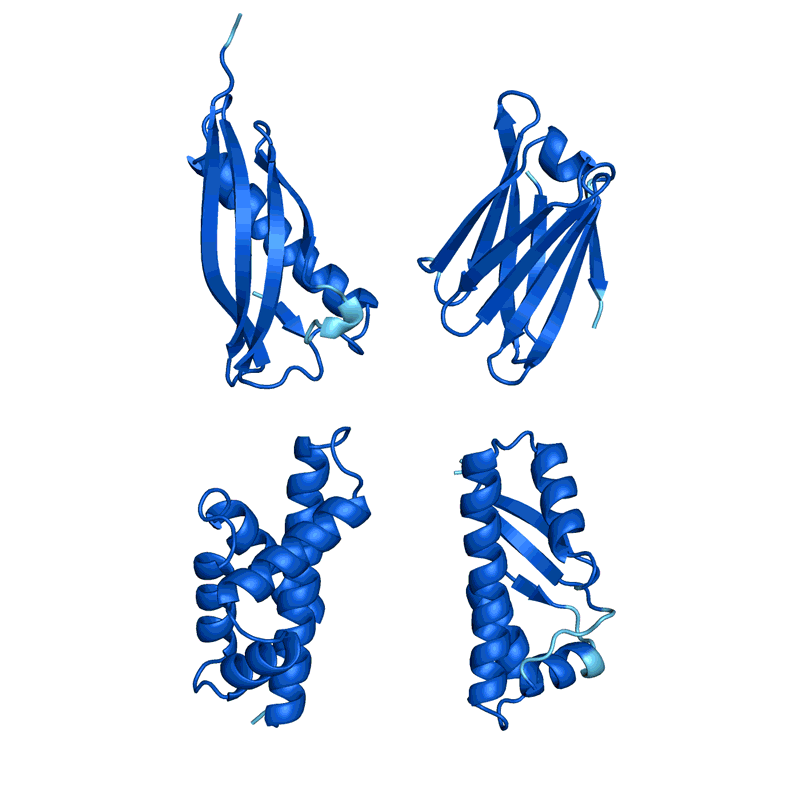
蛋白質(zhì)“幻覺”的四個例子。在每種情況下,AlphaFold都會顯示一個隨機氨基酸序列,預測結構并更改序列,直到軟件有把握地預測它將折疊成具有明確3D形狀的蛋白質(zhì)。顏色顯示預測置信度(從紅色表示非常低的置信度,通過黃色和淺藍色到深藍色表示非常高的置信度)。
破解AlphaFold:其工作原理如何?
當DeepMind發(fā)布其AlphaFold代碼后,Ovchinnikov想要更好地了解該工具的工作原理。幾天之內(nèi),他和包括Steinegger在內(nèi)的計算生物學同事建立了一個名為ColabFold的網(wǎng)站,允許任何人向AlphaFold或RoseTTAFold提交蛋白質(zhì)序列并獲得結構預測。Ovchinnikov設想他和其他科學家會使用ColabFold來嘗試“破解”AlphaFold,例如,通過提供有關目標蛋白質(zhì)序列進化親屬的虛假信息,Ovchinnikov希望他能夠確定網(wǎng)絡是如何學會如此出色地預測結構。
事實證明,大多數(shù)使用ColabFold的研究人員只是想獲得蛋白質(zhì)結構。也有其他人將其用作修改AlphaFold的輸入以處理新應用程序的平臺。“我沒想到會有各種類型的黑客攻擊。”Jumper說。
到目前為止,最流行的黑客攻擊是在蛋白質(zhì)復合物上使用該工具,該復合物由多個相互作用的(通常是相互交織的)肽鏈組成。就像核孔復合物一樣,細胞中的許多蛋白質(zhì)在與多個蛋白質(zhì)亞基形成復合物時發(fā)揮作用。
AlphaFold旨在預測單個肽鏈的形狀,其訓練完全由此類蛋白質(zhì)組成。但該網(wǎng)絡似乎已經(jīng)了解了一些關于復合物如何折疊在一起的知識。AlphaFold的代碼發(fā)布幾天后,東京大學的蛋白質(zhì)生物信息學家Yoshitaka Moriwaki在推特上表示,如果將兩個蛋白質(zhì)序列與長連接序列縫合在一起,它可以準確預測它們之間的相互作用。Baek很快分享了另一個從開發(fā)RoseTTAFold中收集到的預測復合物的技巧。
ColabFold后來加入了預測復合物的能力。同時,在2021年10月,DeepMind發(fā)布了一個名為AlphaFold-Multimer 8的更新,與其前身不同,該更新專門針對蛋白質(zhì)復合物進行訓練。Jumper的團隊將其應用于蛋白質(zhì)數(shù)據(jù)庫中的數(shù)千個復合物,發(fā)現(xiàn)它預測了大約70%的已知蛋白質(zhì)間相互作用。
這些工具已經(jīng)在幫助研究人員發(fā)現(xiàn)潛在的新蛋白質(zhì)伙伴。Elofsson的團隊使用AlphaFold預測了6.5萬個人類蛋白質(zhì)對的結構,根據(jù)實驗數(shù)據(jù)這些蛋白質(zhì)對被懷疑有相互作用。Baker領導的一個團隊使用AlphaFold和RoseTTAFold來模擬酵母編碼的幾乎每對蛋白質(zhì)之間的相互作用,識別出100多個以前未知的復合物。
Elofsson說,這只是起點,他們在預測某些蛋白質(zhì)配對方面做得很好,特別是那些穩(wěn)定的,但很難識別出更短暫的相互作用。“因為它看起來不錯并不意味著它是正確的,”Elofsson說,“你需要一些實驗數(shù)據(jù)來證明你是對的。”
Kosinski認為,核孔復合體的工作是預測和實驗數(shù)據(jù)如何協(xié)同工作的一個很好的例子,“這并不是說我們將所有30種蛋白質(zhì)都放入AlphaFold中,然后把結構拿出來。”為了將預測的蛋白質(zhì)結構放在一起,研究小組使用了核孔復合物的3D圖像,這些圖像是使用一種稱為低溫電子斷層掃描的冷凍電子顯微鏡拍攝的。在一個例子中,可以確定蛋白質(zhì)接近度的實驗在復合物的兩個成分之間產(chǎn)生了令人驚訝的相互作用,AlphaFold的模型隨后證實了這一點。
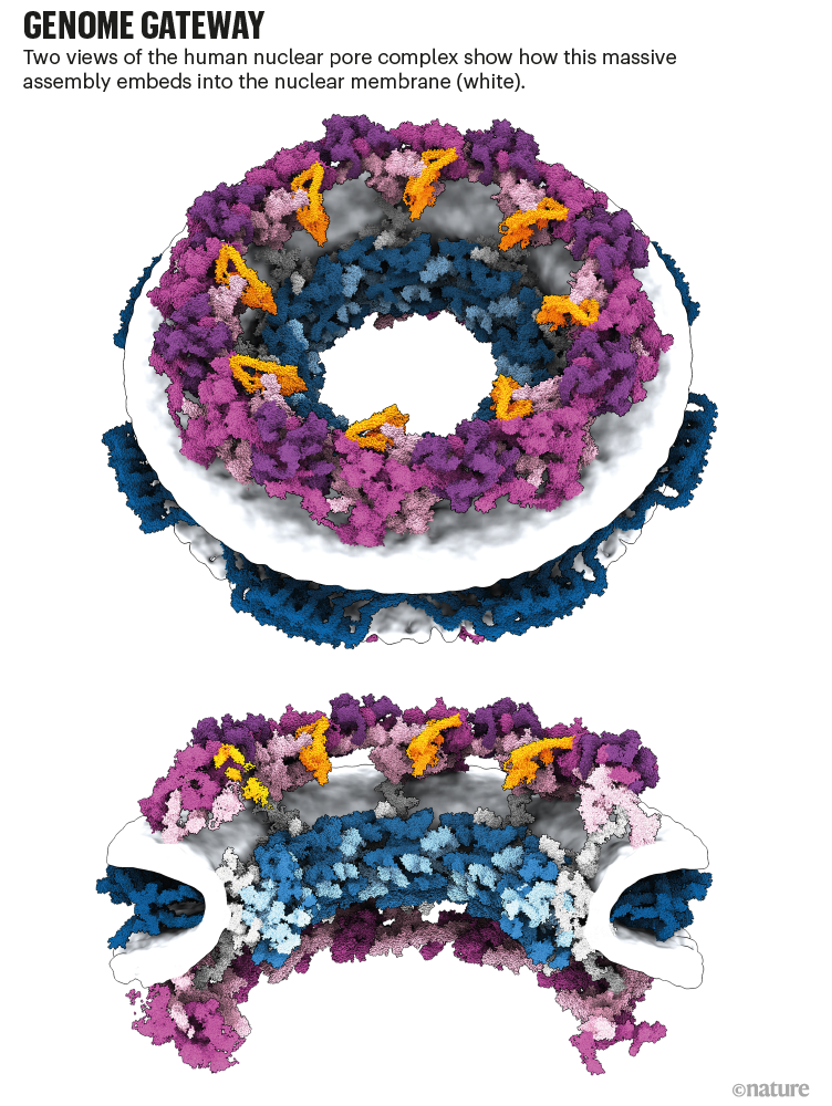
基因組網(wǎng)關:人類核孔復合體的兩種視圖顯示它如何嵌入核膜
??
AlphaFold的限制:預測蛋白質(zhì)新突變的后果
對于AlphaFold取得的所有進展,科學家們表示,重要的是要清楚它的局限性,特別是因為不專門預測蛋白質(zhì)結構的研究人員也在使用它。
嘗試將AlphaFold應用于破壞蛋白質(zhì)自然結構的各種突變,包括與早期乳腺癌相關的突變,已被證實該軟件無法預測蛋白質(zhì)新突變的后果,因為沒有進化相關的序列可供檢查。
AlphaFold團隊現(xiàn)在正在考慮如何設計神經(jīng)網(wǎng)絡來處理新的突變。Jumper預計這將需要網(wǎng)絡更好地預測蛋白質(zhì)如何從展開狀態(tài)變?yōu)檎郫B狀態(tài)。哥倫比亞大學計算生物學家Mohammed AlQuraishi說,這可能需要僅依靠它所學到的蛋白質(zhì)物理學知識即可預測結構的軟件。“我們感興趣的是,在不使用進化信息的情況下從單個序列進行預測,”他說,“這是一個關鍵問題,但仍然懸而未決。”
AlphaFold還旨在預測單個結構。但是許多蛋白質(zhì)具有多種構象,這對其功能可能很重要。“AlphaFold不能真正處理可以采用不同構象的不同結構的蛋白質(zhì)。”Schueler-Furman說。預測是針對孤立結構的,而許多蛋白質(zhì)與配體(如DNA和RNA)、脂肪分子和礦物質(zhì)(如鐵)一起發(fā)揮作用。“我們?nèi)匀蝗鄙倥潴w,我們?nèi)鄙訇P于蛋白質(zhì)的其他一切。”Elofsson說。
AlQuraishi認為,開發(fā)這些下一代神經(jīng)網(wǎng)絡將是一個巨大的挑戰(zhàn)。AlphaFold依賴于數(shù)十年的研究,這些研究產(chǎn)生了網(wǎng)絡可以學習的蛋白質(zhì)實驗結構。目前無法獲得如此大量的數(shù)據(jù)來捕捉蛋白質(zhì)動力學,或者蛋白質(zhì)可以與之相互作用的數(shù)萬億個小分子的形狀。Jumper補充說,蛋白質(zhì)數(shù)據(jù)庫包括蛋白質(zhì)與其他分子相互作用時的結構,但這僅捕獲了一小部分化學多樣性。
研究人員認為,他們需要時間來確定如何最好地使用AlphaFold和相關的人工智能工具。AlQuraishi看到了電視發(fā)展早期的相似之處,當時一些節(jié)目由廣播電臺組成,只是閱讀新聞,“我認為我們將找到我們尚未設想的結構的新應用。”
AlphaFold革命的終點在哪里,誰也說不準。“事情變化太快了,”Baker說,“即使是下一年,我們也將看到使用這些工具取得的重大突破。”EMBL-EBI的計算生物學家Janet Thornton認為,AlphaFold的最大影響之一可能只是說服生物學家對計算和理論方法的見解更加開放,“對我而言,革命就是思維方式的改變”。
AlphaFold革命激發(fā)了Kosinski的遠大夢想,他認為受AlphaFold啟發(fā)的工具不僅可用于對單個蛋白質(zhì)和復合物進行建模,還可以對整個細胞器甚至細胞進行建模,直至單個蛋白質(zhì)分子的水平,“這是我們未來幾十年的夢想。”



- 報料熱線: 021-962866
- 報料郵箱: news@thepaper.cn
互聯(lián)網(wǎng)新聞信息服務許可證:31120170006
增值電信業(yè)務經(jīng)營許可證:滬B2-2017116
? 2014-2025 上海東方報業(yè)有限公司

